Advertise With UsAdvertise on our extensive network of industry websites and newsletters.
Advertise With UsAdvertise on our extensive network of industry websites and newsletters.
 Get the PBR newsletterSign up to our free email to get all the latest PBR
news.
Get the PBR newsletterSign up to our free email to get all the latest PBR
news.Vertex Pharmaceuticals (Europe) has received a positive opinion from the European Medicines Agency’s (EMA) Committee for Medicinal Products for Human Use (CHMP) for Kalydeco (ivacaftor) to treat people with cystic fibrosis (CF) aged 12 to
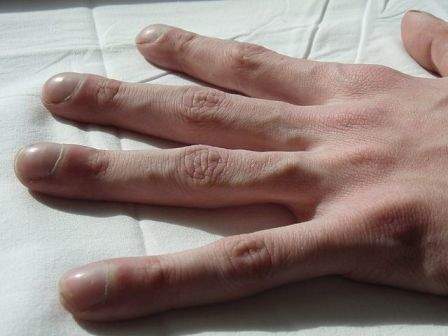
Image: Clubbing in the fingers of a person with cystic fibrosis. Photo: courtesy of Jerry Nick, M.D.
Subscribe to our email newsletter
The committee has recommended Kalydeco to treat people with CF aged 12 to <24 months who have at least one of the following nine mutations in their cystic fibrosis transmembrane conductance regulator (CFTR) gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N or S549R.
If the European Commission issues a favorable adoption of the EMA CHMP opinion for the extension of indication, ivacaftor will be the first and only medicine approved in Europe to treat the underlying cause of CF in patients aged 12 to <24 months, who have specific mutations in the CFTR gene.
Vertex chief medical officer and global medicines development and medical affairs executive vice president Dr Reshma Kewalramani said: “Cystic fibrosis is a chronic, progressive disease that is present at birth, with symptoms often occurring in infancy, so early treatment is crucial to deliver the best possible outcomes for patients.
“Today’s announcement marks an important step towards allowing young CF patients to benefit from treatment at an early stage of their disease, and brings us one step closer to our goal of treating all people living with CF.”
The submission was supported by data from the ongoing Phase 3 open-label safety study (ARRIVAL) of children with CF aged 12 to <24 months who have one of 10 mutations in the CFTR gene that demonstrated a safety profile consistent with that observed in previous Phase 3 studies of older children and adults, and improvements in sweat chloride, a key secondary efficacy endpoint.
Ivacaftor is already approved in Europe for the treatment of CF in patients aged two years and older who have one of the nine following mutations in the CFTR gene: G551D, G1244E, G1349D, G178R, G551S, S1251N, S1255P, S549N or S549R.
It is also approved for the treatment of CF in patients aged 18 years and older who have an R117H mutation in the CFTR gene.
Cystic fibrosis is a rare, life-shortening genetic disease affecting approximately 75,000 people in North America, Europe and Australia.
The ARRIVAL study is an ongoing Phase 3 open-label safety study of 25 children with CF aged 12 to <24 months who have one of 10 mutations in the CFTR gene (G551D, G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P, G1349D or R117H; patients with the R117H mutation were eligible to enroll in regions where ivacaftor is approved for use in patients 2 through 5 years of age with an R117H mutation).
The study demonstrated a safety profile consistent with that observed in previous Phase 3 studies of older children and adults; most adverse events were mild or moderate in severity, and no patient discontinued due to adverse events.
Treatment was interrupted in two patients who had elevated liver enzymes greater than eight times the upper limit of normal, but continued to receive ivacaftor after a dose interruption. The most common adverse events (≥30%) were cough (74%), pyrexia (37%), elevated aspartate aminotransferase (37%), elevated alanine aminotransferase (32%) and runny nose (32%). Four serious adverse events were observed in two patients.
Kalydeco (ivacaftor) is the first medicine to treat the underlying cause of CF in people with specific mutations in the CFTR gene.
Known as a CFTR potentiator, ivacaftor is an oral medicine designed to keep CFTR proteins at the cell surface open longer to improve the transport of salt and water across the cell membrane, which helps hydrate and clear mucus from the airways.